MRI visualisation of drug delivery/release
The design of imaging procedures aimed at providing pharmacologists/ clinicians a valuable in vivo and minimally-invasive support to visualize the effective delivery and release of a drug in the diseased region is very crucial to improve the efficiency of a pharmacological therapy and to optimize the therapeutic planning on an individual base (personalized medicine). This research area, which is part of theranosis, requires the development of chemicals that have to generate an imaging response as a function of the delivered and/or released drug. In principle, imaging protocols for the visualization of drug delivery can be designed for almost all the available imaging modalities (nuclear, CT, optical, US, MRI, and hybrid technologies). However, for imaging drug release purposes, MRI is certainly the choice of election because of the widespread and successful preclinical and clinical use, the good spatio-temporal resolution, the possibility to reach deep tissues/organs without any limitations, and the rich portfolio of agents and contrast modalities available. The motivation of using nanocarriers in the pharmacological field is mainly driven by the necessity to improve the therapeutic index of a drug. The rational is to influence the biodistribution of the drug to favour (by passive or active targeting) the accumulation and availability at the target organ, thereby improving therapeutic efficacy and reducing side effects. However, to exert the effect, the drug needs to be released from the carrier. For the nanomedicines currently approved for clinical use, this fundamental step occurs spontaneously, i.e. following the natural degradability of the nanocarrier interacting with tissue components. However, a significantly better control of the release can be achieved through a specific stimulation, especially suitable for treating solid tumours. The release of a drug is dependent on many factors, including the physico-chemical properties of the nanocarrier, and therefore, the development of new drug delivery systems is still an important research area.
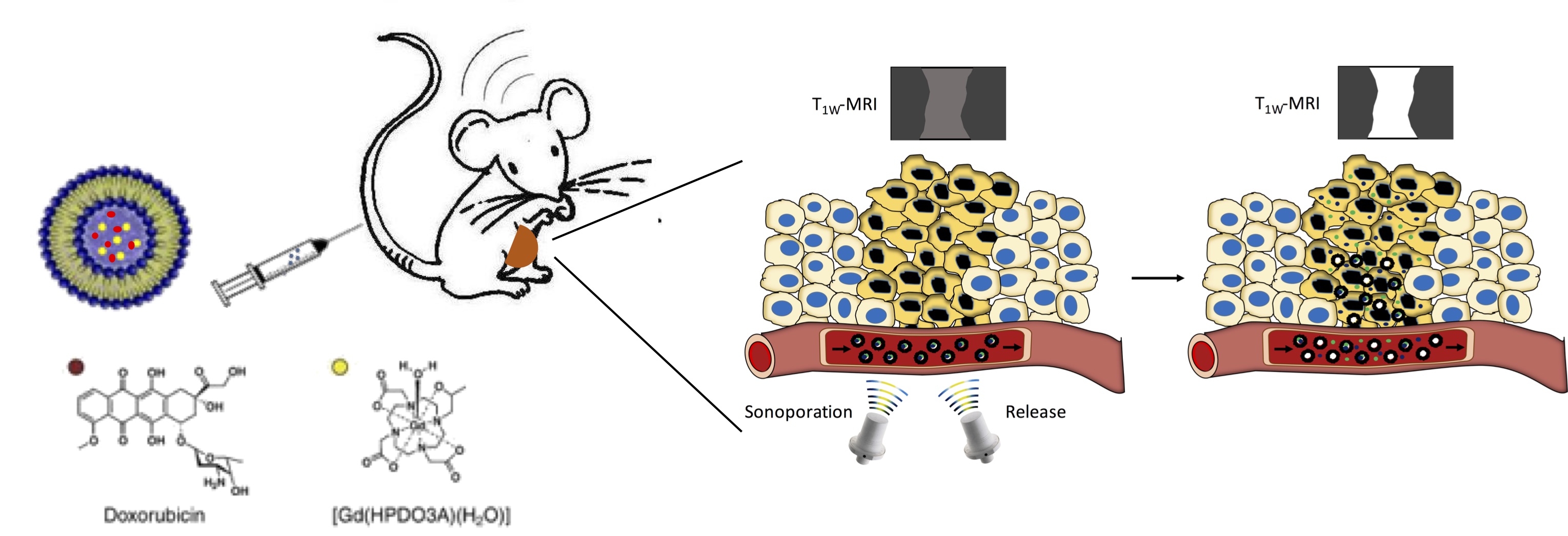
MRI visualization of the release of doxorubicin from liposomes stimulated by local application of ultrasound
The release of a drug from a nanocarrier can be stimulated by several factors, endogenous (e.g. pH, enzyme), or externally applied (heat, light, US ...). It has been demonstrated that liposomes can release their content upon stimulation with pulsed low intensity non-focused US (pLINFU), which can be broadly defined as pulsed, planar, acoustic waves with intensity lower than 10 W/cm2 and US frequencies ranging from low (20 kHz) to therapeutic (1-3 MHz) frequency. The lower energy associated with pLINFU produces minimal or no thermal effects and the release of the drug mainly results from the mechanical interaction between the acoustic waves and the nanocarrier. A practical approach to visualize by MRI the release of the drug from liposomes consists of encapsulating a hydrophilic paramagnetic agent (based on Gd3+ or Mn2+ ions) in the aqueous inner cavity of the nanovesicle. Upon the entrapment, the MRI contrast is "silenced" and its activity is recovered when the agent is released.
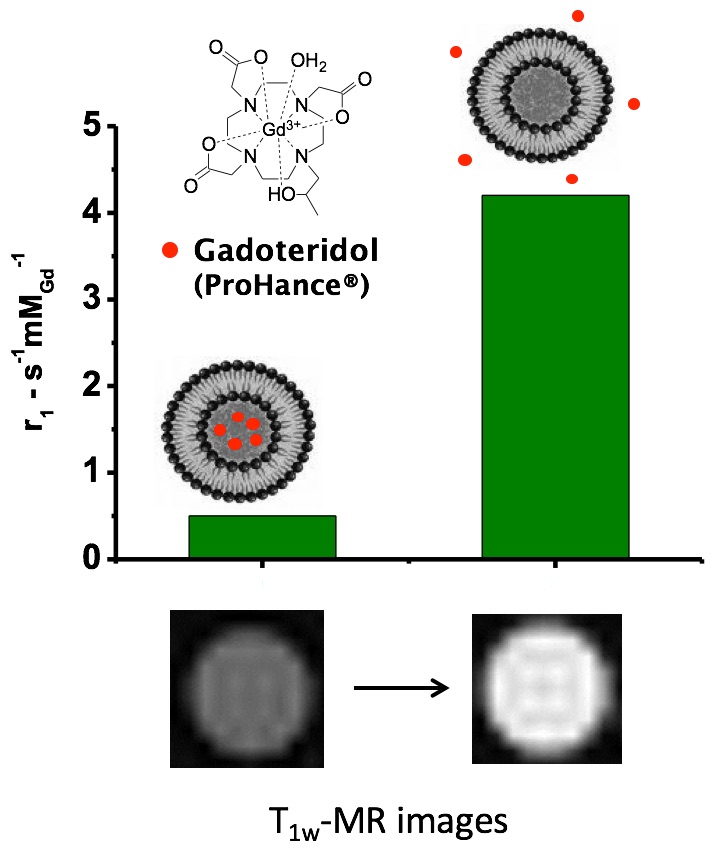
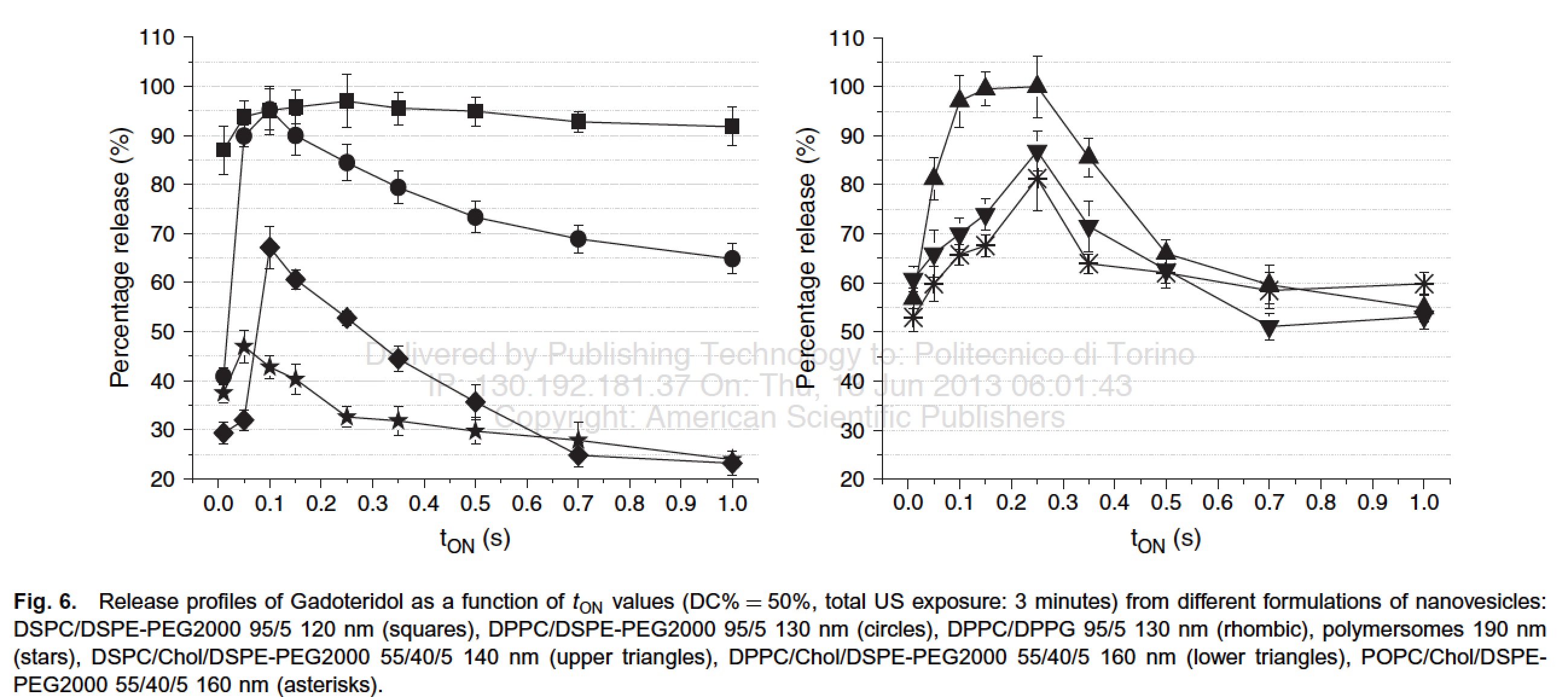
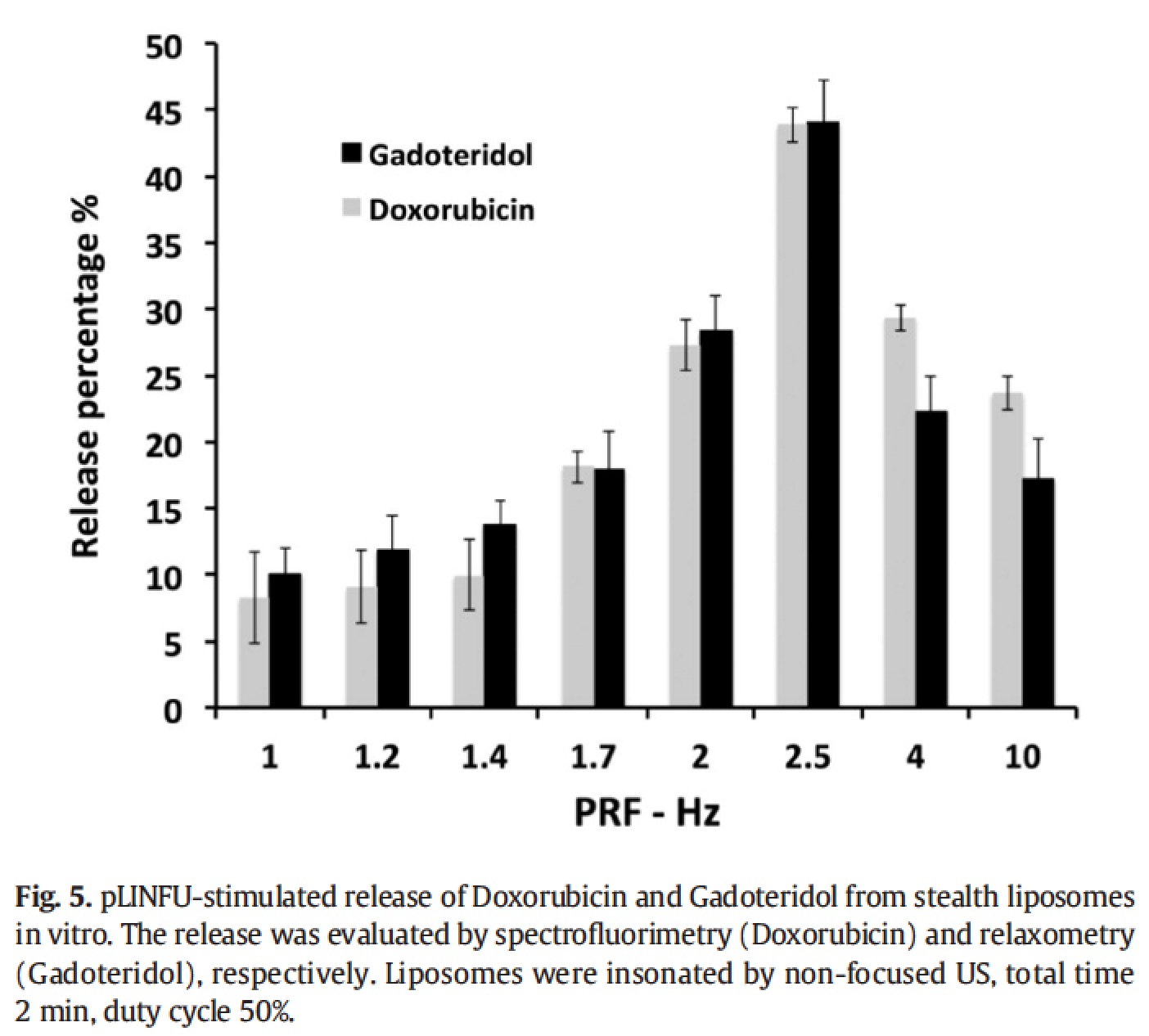
In the first paper [Giustetto P. et al. 2013] , it has been demonstrated that the release of the imaging probe is strongly dependent on the pulse repetition frequency of the insonation (main US frequency 27 kHz) and it is even affected by subtle changes in the chemical composition of the nanocarrier.
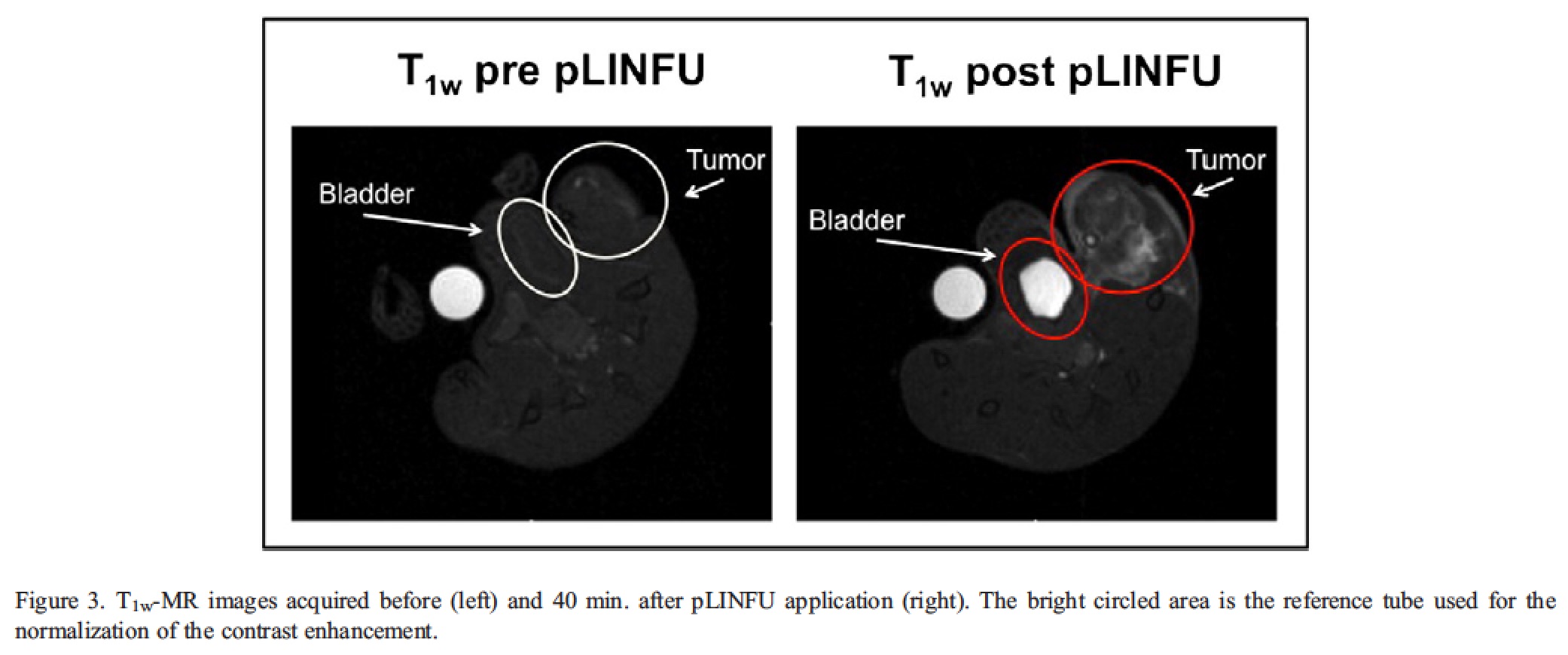
A step forward was achieved later on [Rizzitelli S. et al. 2014] , when the US stimulation (3 MHz) was applied in vivo on a subcutaneous melanoma mouse tumour. In addition to the contrast enhancement observed in the tumour after the US stimulation, a strong evidence about the effective release of the agent was gained by the detection of the T1 contrast enhancement in kidneys and bladder. In fact, since gadoteridol has a rapid renal excretion (t1/2 of ca. 3 hours in mice), the higher contrast observed in both organs for the Gd+/US+ group supports the remote release of the agent.
To test the therapeutic potential of this approach, liposomes were co-encapsulated with the drug doxorubicin and tested on a mouse model of breast cancer [Rizzitelli S. et al. 2015] . First of all, it was checked that the release of the two components (the imaging probe and the drug) were similar in order to consider the contrast enhancement as a good predictor for the drug release.
The results confirmed the strong dependence of the release on the pulse repetition frequency, and, very important, highlighted the close similarity in the release of the two compounds over the entire range investigated.
The planning of the experiment is reported below:
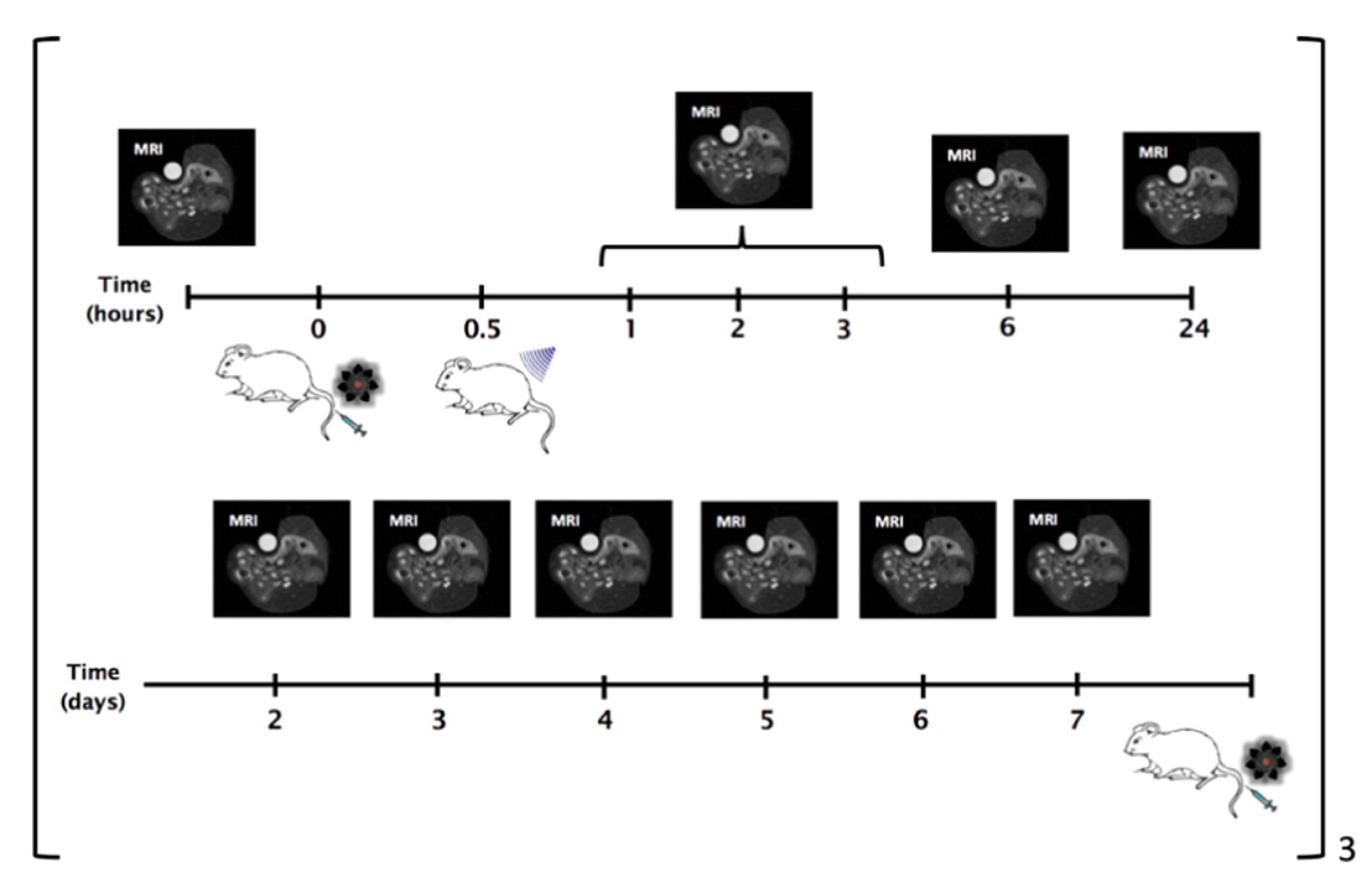
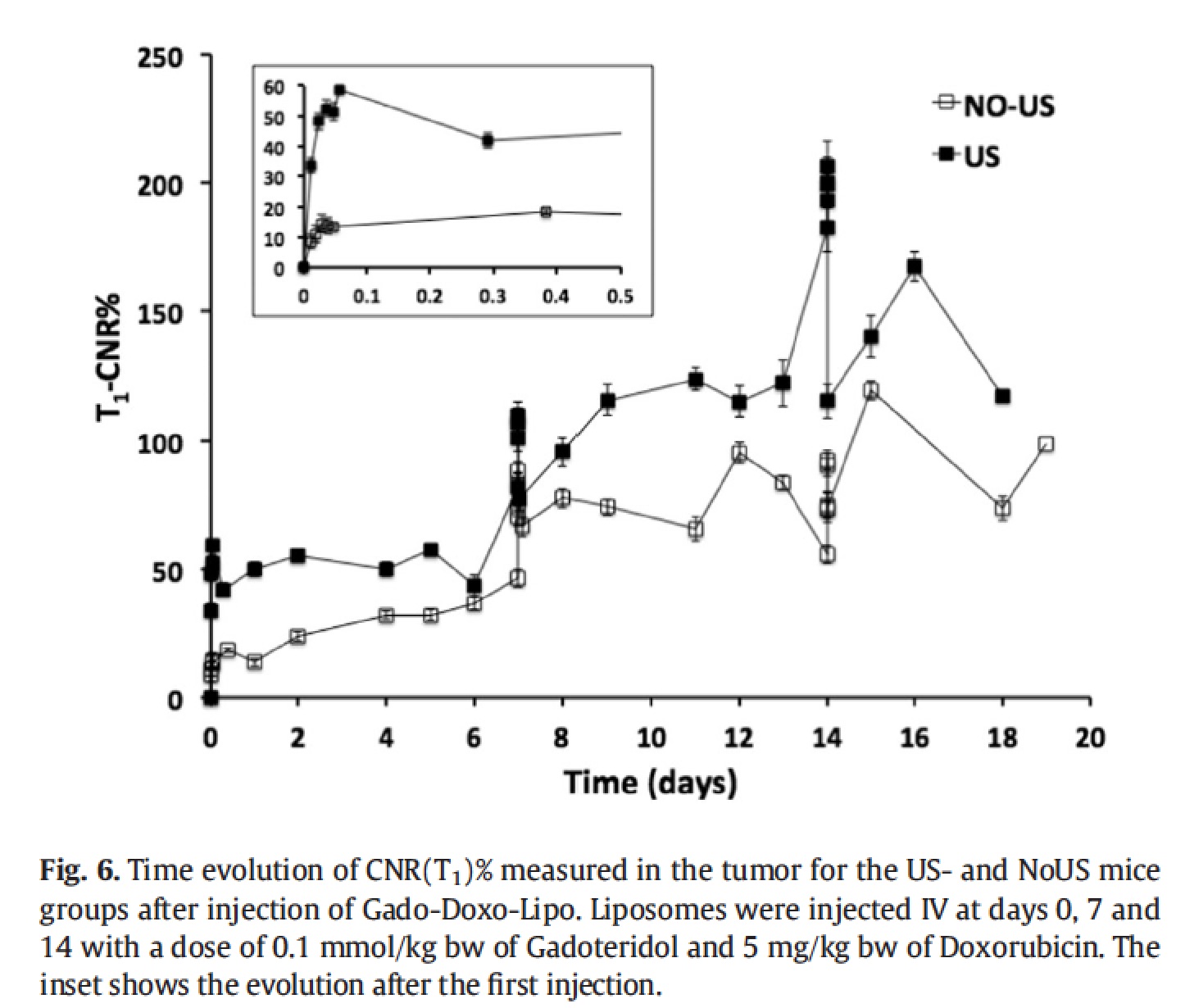
The liposomes were administered once a week for three weeks and the US stimulation was locally applied after each administration. MRI were carried out more frequently the day of the administration and then once a day. pLINFU-stimulated mice showed significantly higher T1 contrast than the untreated group, as expected in case of successful release of the MRI agent. The enhancement for the US-group was maximal just after the stimulation and decreased within 6 h. Contrarily, a much smaller enhancement was detected in the NoUs-Group, which was due to the intratumour circulation of the intact "MRI-quenched" liposomes.
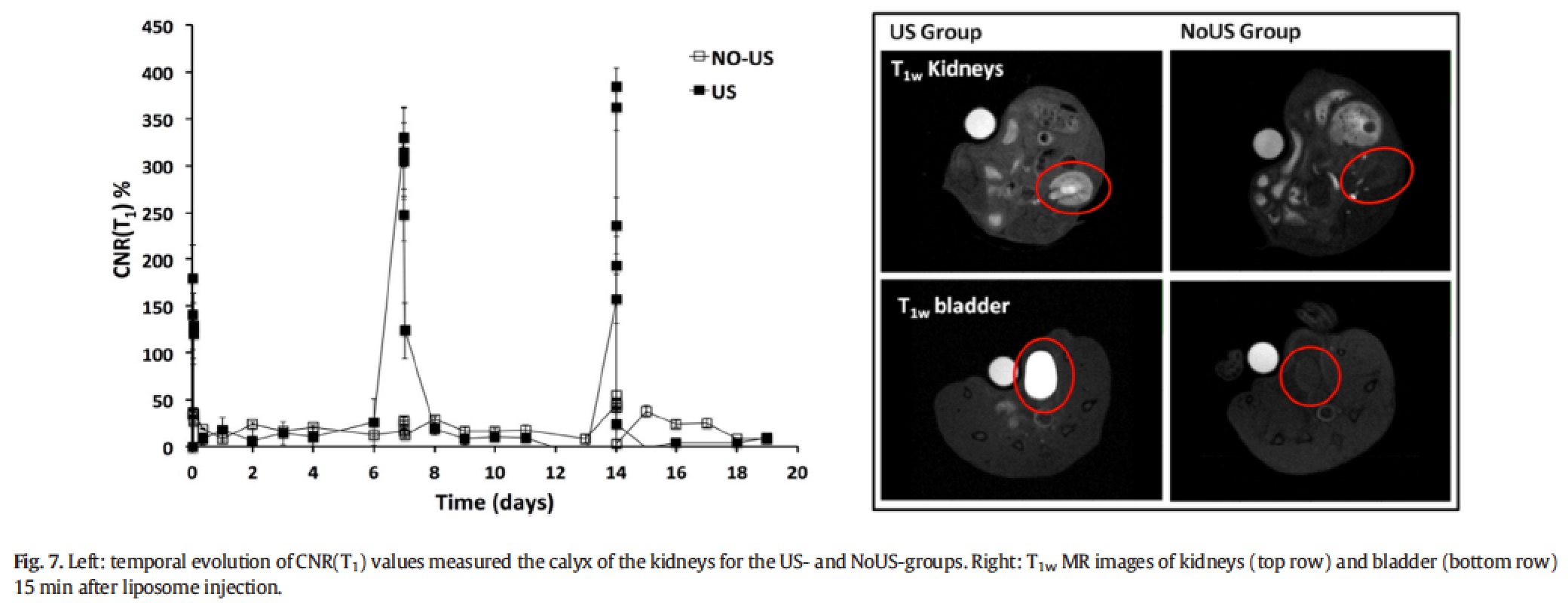
As expected from the renal excretion of the MRI agent, the release of the agent in the tumour is associated with the accumulation of the probe in the kidney calyx and bladder. The presence of a very bright T1 contrast in both of these compartments just after the tumour insonation was a clear evidence of the effective intratumour release of the MRI probe triggered by the local pLINFU application.
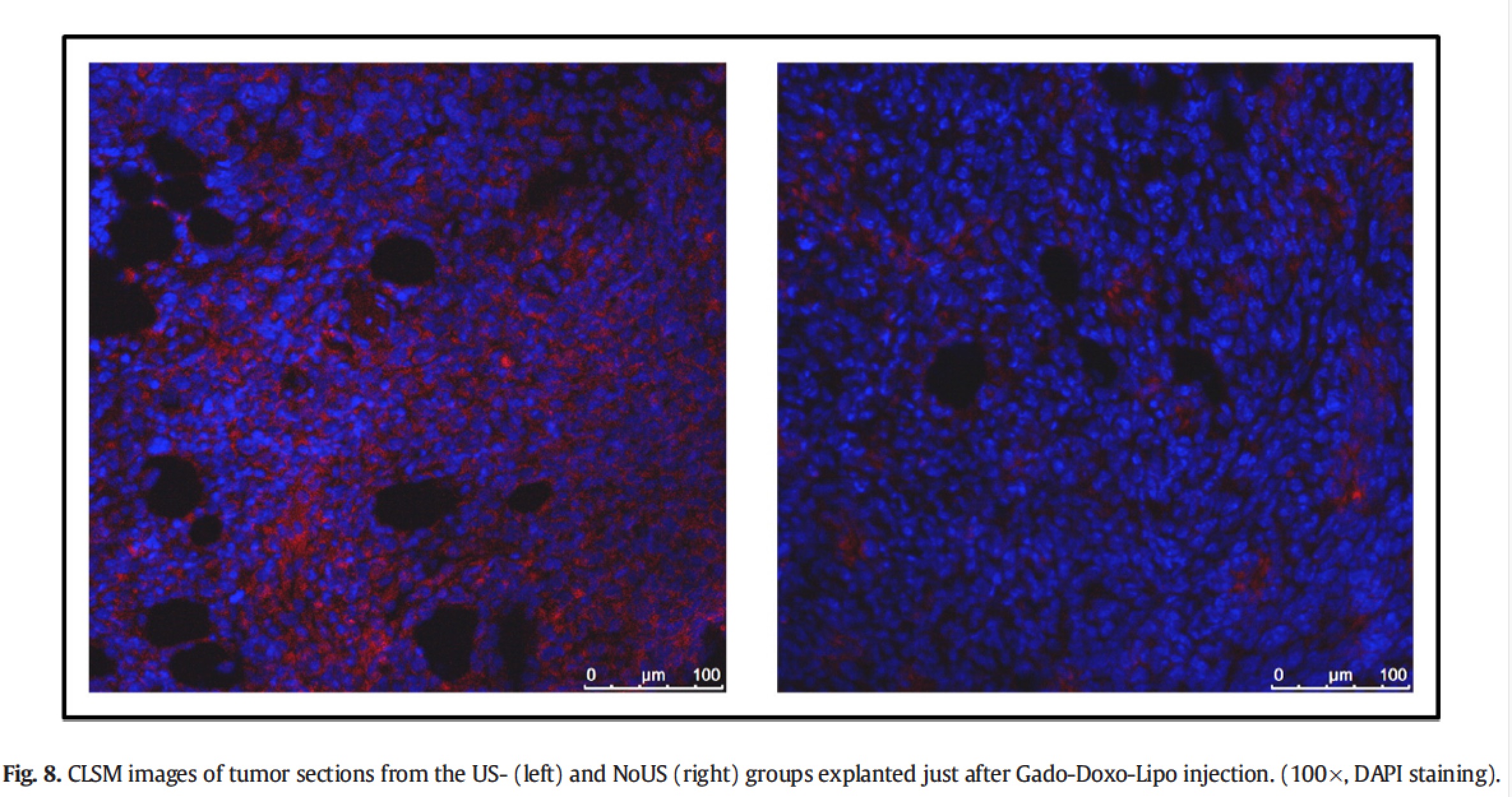
As further proof of the effective release of doxorubicin in the tumour stimulated by pLINFU, confocal fluorescence microscopy slices of tumour treated showed a diffuse red fluorescence form the drug, whereas in the untreated tumour the fluorescence was much more localized.
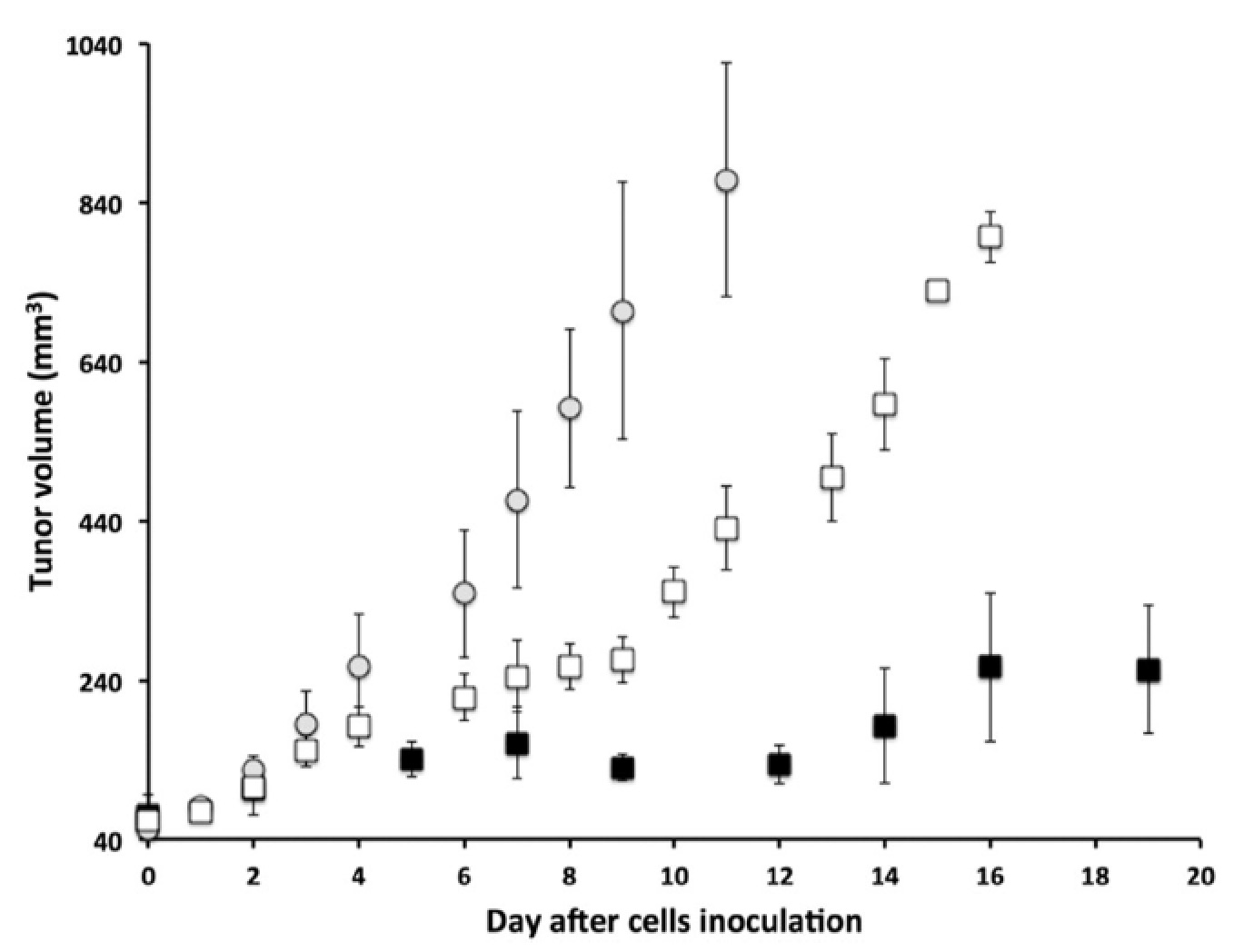
Importantly, the tumour of the mice stimulated with pLINFU after three weeks of treatment grew significantly less with respect to the untreated animals and the controls (that did not receive liposomes).
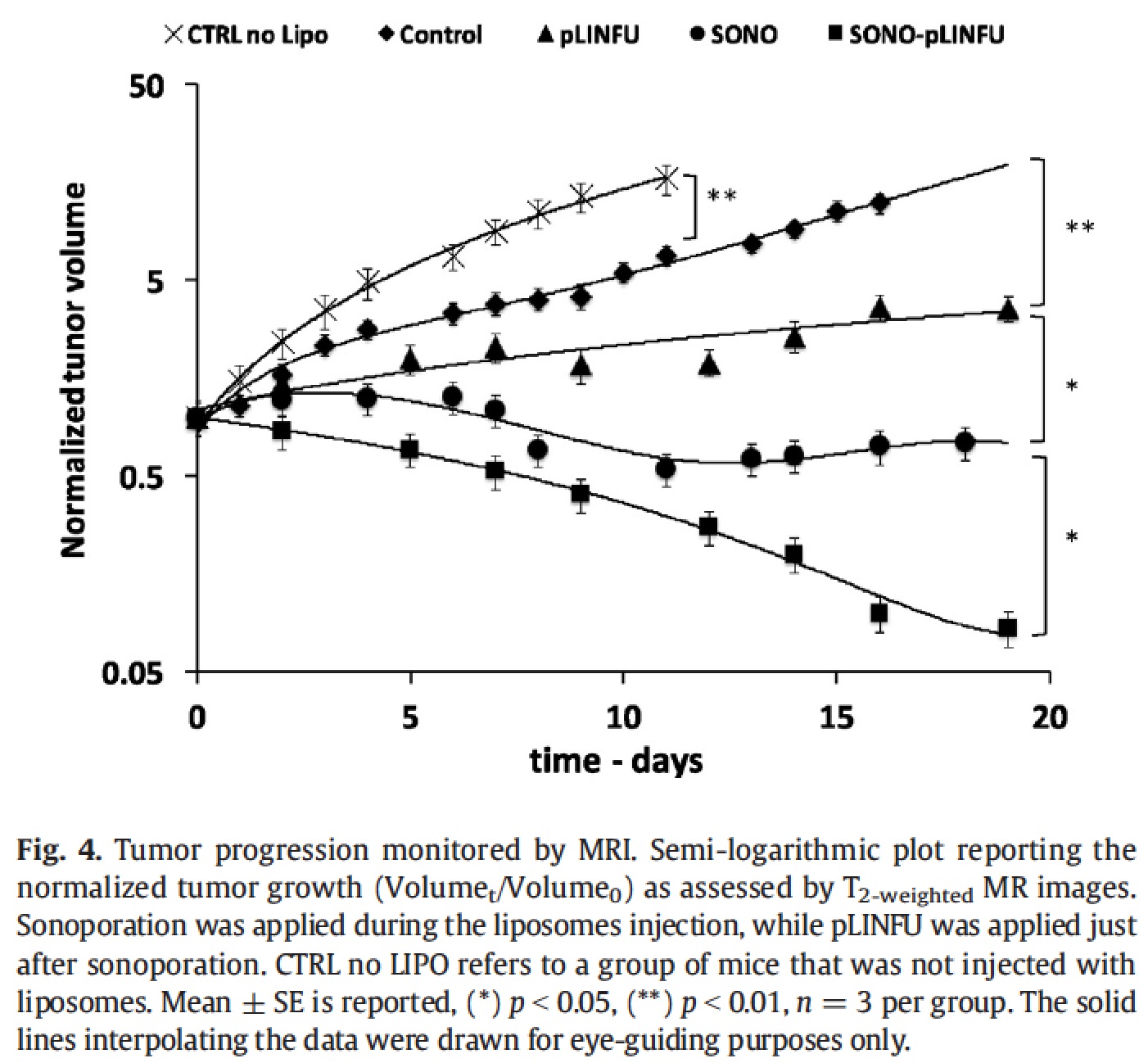
A further improvement in the therapeutic outcome was achieved by adding a second acoustic stimulus before that one applied for triggering the release of the drug [Rizzitelli S. et al., 2016] . This second pulse was designed to induce sonoporation, i.e. to permeabilize cell membrane with the aim of increasing the total amount of drug that diffuses in the tumour. The gain in the therapeutic performance of this improved method was excellent, and the combination between release and sonoporation led to a complete remission of the tumour after the three weeks of treatment.
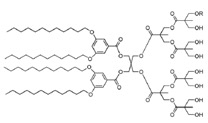
Among the new nanocarriers explored for improving the imaging and pharmacokinetic properties, dendrimersomes have displayed interesting results. Dendrimersomes are self-assembling nanovesicular systems consisting of a double-layer membrane made up of amphiphilic dendrimers like that one shown below:
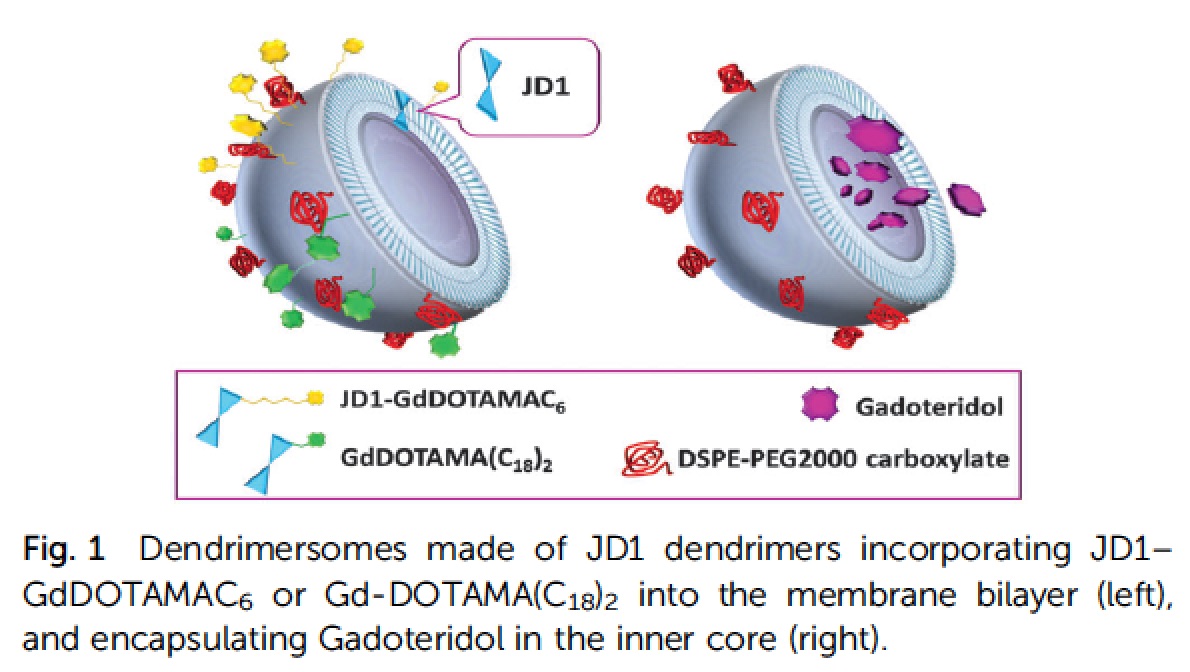
Filippi et al. 2014 demonstrated the versatility of dendrimersomes to be loaded with MRI agents either hydrophilic (Gadoteridol) or amphiphilic (Gd-DOTAMA(C18)2). The relaxivity of the system loaded with the amphiphilic complex was similar to liposomes, whereas the relaxivity of the system loaded with Gadoteridol was higher than that observed for conventional liposomes, demonstrating the high water permeability of the dendrimersomes membrane. The stability and biocompatibility of a series of four dendrimersomes consisting of two amphiphilic dendrimers reported in literature and two lower-generation amphiphilic dendrimers synthesized ex-novo (in collaboration with prof. L. Tei, UniUPO) were compared in detail by Filippi et al. 2015 . The new dendrimers were able to form dendrimersomes with low polydispersion and highest stability. Furthermore, no signs of cytotoxicity and changes in proliferation rate were observed even after 48 hours of incubation with different cell lines (RAW 264.7, J774.A1 and NIH/3T3). Finally, the more stable formulation of dendrimersomes was injected into a healthy mouse (first in vivo experiments for this class of nanocarriers) to evaluate plasma half-life. The value obtained, around 70-80 min, was very similar to that one obtained injecting conventional liposomes.